Shwachman-Diamond综合征合并急性双系列白血病1例
患者男性,16岁2个月,体型矮小。2020年11月因乏力、发热伴鼻出血7个月就诊北京大学第三医院血液科。
2020年4月患者无明显诱因出现乏力伴发热,无明显感染症状,合并鼻腔出血。2020年10月就诊外院考虑为急性B淋巴细胞白血病(B-ALL),予VDCLP方案(长春地辛+去甲氧柔红霉素+环磷酰胺+门冬酰胺酶+泼尼松)1次。入院后血常规示白细胞3.52×109/L,中性粒细胞1.74×109/L,血红蛋白136 g/L,血小板62×109/L,进行外院骨髓病理、流式细胞术、形态会诊均为两类幼稚细胞,最终诊断为急性双系列白血病[B-ALL/急性髓系白血病(AML)]。患者化疗后骨髓抑制期长而多次延长化疗间期,化疗期间反复发热伴粒细胞缺乏(感染累及肝脏、肺、肛周),血培养及二代基因测序结果为白念珠菌感染。患者是无血缘关系父母的第2个孩子,足月出生,出生体重大致正常,入院时身高156 cm(同年龄组<3%),体重39 kg,生长激素1.24 μg/L(正常值<3 μg/L);其哥哥身材矮小,血常规提示全血细胞减少(因拒绝行骨髓穿刺检查,血细胞减少原因不明)。
骨髓穿刺及活检:外院行第1次骨髓穿刺,形态为原始细胞62.5%,骨髓发育不良;流式细胞术报告该群细胞表达CD34、CD13、CD33、CD38、人类白细胞相关抗原(HLA)-DR,部分表达CD10、CD19、CD117、cCD22,不表达CD2、CD3、CD5、CD7、CD8、CD11b、CD14、CD15、CD16、CD20、CD56、CD64、髓过氧化物酶(MPO),考虑急性淋巴细胞白血病(ALL)/淋巴母细胞淋巴瘤;细胞遗传学为复杂核型:44,XY,der(5,5)(q10;q10),add(14)(p11),-15,der(17)t(1;17)(q32;q24),-20,-21,add(21)(q22),-22,+3mar[3]/46,XY[17];分子生物学发现TP53基因p.R248Q热点突变69.6%;骨髓病理提示B-ALL。后经本院形态会诊见幼稚淋巴细胞20%,幼稚单核细胞20%,原始细胞10%(图1),病理会诊发现MPO阳性。行VDCLP方案诱导化疗1周期复查骨髓穿刺,形态提示骨髓增生活跃,未见原始淋巴细胞;流式细胞术未见幼稚细胞;细胞遗传学恢复为正常核型;TP53突变频率下降至54.2%。影像学检查:骨密度提示骨质疏松症,X线片提示干骺端发育不良。腹部CT及MR提示肝脏多发结节,不除外肝脏脓肿,胰腺重度脂肪化,无肝脾肿大。胸CT提示双肺多发结节,磨玻璃影改变。
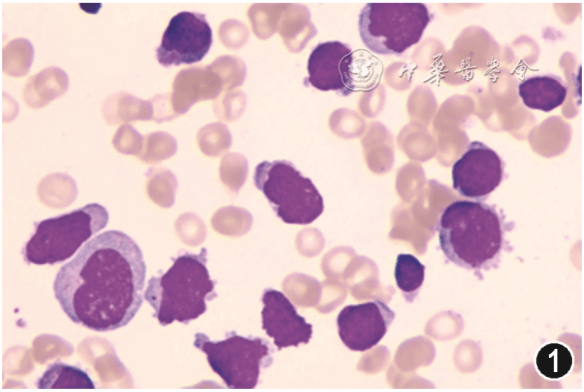
图1 急性双系列白血病患者骨髓涂片可见原始细胞、幼稚淋巴细胞、幼稚单核细胞 瑞氏染色 ×1000
基因检测:患者外周血进行全外显子测序,结果为Shwachman-Bodian-Diamond综合征(SBDS)基因存在2个杂合突变。第一,c.258+2T>C突变是有明确致病意义,主要起到氨基酸剪切突变,也对氨基酸错义突变起一定作用,在千人基因组数据中频率为0.001 597。经过家庭分析及验证,患者父亲和哥哥为杂合突变,母亲无该位点突变(图2)。第二,c.115T>C突变是与临床表型相关但临床意义未明变异,导致氨基酸错义突变,经过家庭分析及验证,患者母亲和哥哥为杂合突变,父亲无该位点突变(图3)。此外,患者存在TP53基因c.743G>A位点突变,疑似致病且与不良预后独立相关,而其亲属无突变。诊断为Shwachman-Diamond综合征(SDS)。
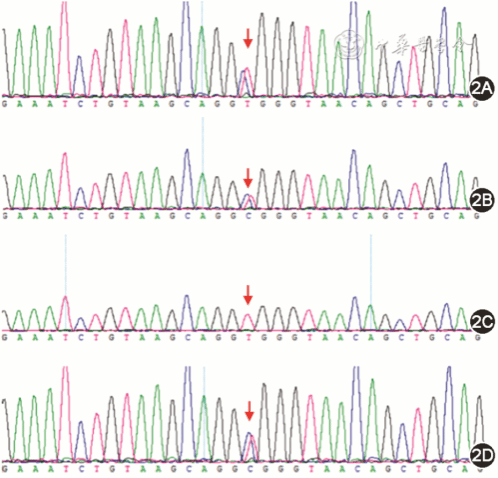
图2 急性双系列白血病患者及家属外周血全外显子测序Shwachman-Bodian-Diamond综合征(SBDS)基因突变位点c.258+2T>C(箭头示位置)2A 示患者为突变杂合子;2B 示患者父亲为突变杂合子;2C 示患者母亲无突变;2D 示患者哥哥为突变杂合子
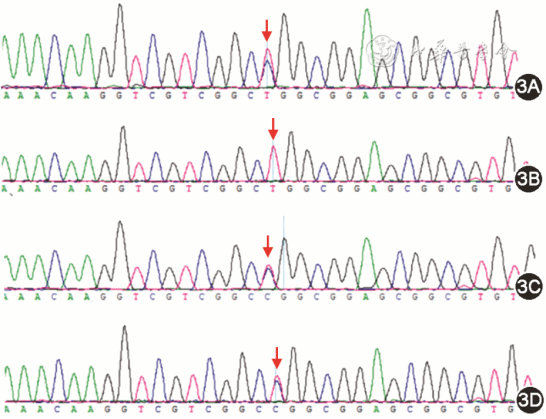
图3 急性双系列白血病患者及家属外周血全外显子测序Shwachman-Bodian-Diamond综合征(SBDS)基因突变c.115T>C位点(箭头示位置)3A 示患者为突变杂合子;3B 示患者父亲为无突变;3C 示患者母亲为突变杂合子;3D 示患者哥哥为突变杂合子
 典型病例
典型病例